
By Kayleigh Gultig
A brief history of genome-sequencing
2001- a breakthrough year for geneticists: the year in which the most important biological code was published. Groups from the US, UK, Japan, Germany and China collaborated to publish the first draft sequence of the human genome [1] based on the sequences of anonymous volunteers. And so, the first pieces in the human genetics puzzle were fitted. Twenty-one years later and a gaping hole in the puzzle glares out at me – the lack of diversity. Why has Africa, the birthplace of humanity, so often been excluded from genetic sequencing efforts? Has this hole been noticed by others and when will it be filled?
With the rapid advancement of technology, sequencing the human genome has become cheaper and easier. This allows for researchers to study the human genome and advance our understanding of biology and medicine. A genome wide association study is a research tool used to identify statistical associations between genes and a trait or disease. Genome wide association studies (GWAS) emerged with the goal of identifying associations between genetic makeup (genotype) and the physical, observable traits resulting from gene expression (phenotype) [2]. GWAS involve the collection of genotypic and phenotypic data from many participants. Unfortunately, GWAS suffer from a Eurocentric bias. As of December 2020, GWAS consisted of 78% European participants [3]. Due to the majority of the available genetic data being from European origin, this is perpetuating the research bias as datasets from other ancestries are frequently deemed too small to warrant inclusion in future research.
How did such a bias emerge? One of the first groups involved in sequencing the human genome was the International Human Genome Sequencing Consortium (IHGSC). This is a publicly funded group of researchers, who worked along with the private biotechnology company known as Celera Genomics. Both groups included participants from diverse ethnic groups, however, the projects were based in the US and researchers were mainly from first world countries. Despite these initial projects achieving a certain level of diversity of participants, the driving force behind the research was more Eurocentric and this ultimately led to some bias in their work. This, together with the high cost and resource demands of genome sequencing left third world countries at an immediate disadvantage.
Thanks to the reference human genome, not every nucleotide in an individual’s genetic code needs to be sequenced. Imputation is the process by which parts of an individual’s genetic sequence are inferred. This is possible due to the non-random association of certain genetic variants in a population [2] known as linkage disequilibrium (LD). In addition to being a tool in genetic research, LD also poses a problem as the pattern of LD is not conserved across populations [4]. For example, while gene x and gene y may be linked in a European population, in an African population, gene x may be linked with gene z. Thus, using gene x for imputation would result in gene y being incorrectly inferred in the African population. This means that the ability for scientists to generalise genetic findings in a European cohort is not as high as initially proposed.
ALS – a clinical example of the importance of more genetic diversity in research
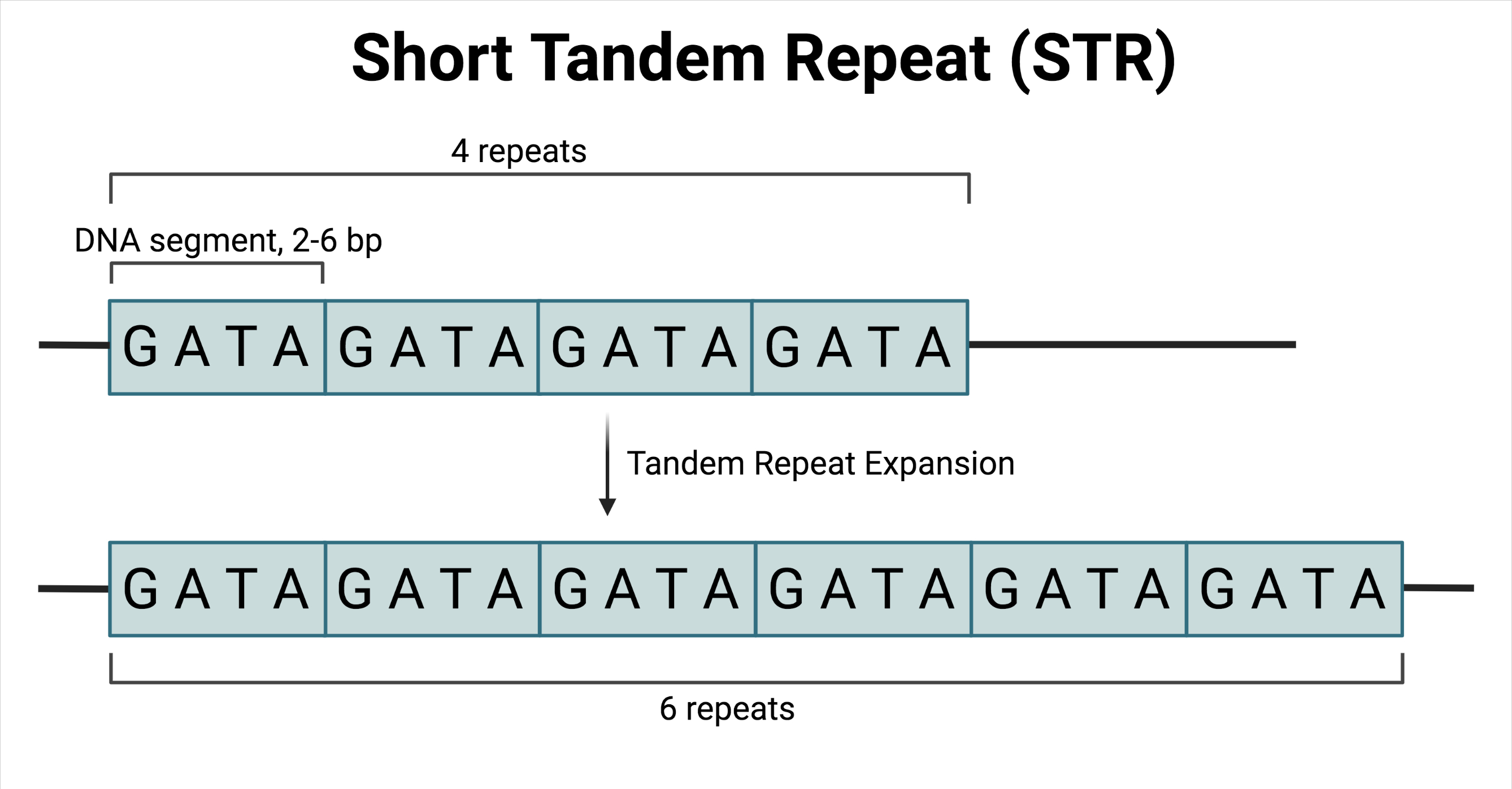
An example of this lack of generalisability is evident in the field of amyotrophic lateral sclerosis (ALS) research. ALS is a fatal neurodegenerative disease characterised by motor neuron loss. In European populations, the most common genetic cause of ALS is an abnormal increase in the number of repeated GGGGCC sequences in the C9ORF72 gene [5]. The GGGGCC sequence is an example of a sequence in the genome that is repeated a variable number of times (Fig 1). Such a sequence is known as a short tandem repeat (STR).

STRs are normal occurrences in the genome, however, the repeat number may expand beyond a critical size known as an expansion mutation [6]. Such mutations are considered disease-causing or pathogenic. In order to define pathogenic repeats, a normal repeat range from a healthy population is required. This is challenging as the frequency distribution of normal repeat sizes varies between populations. In African populations, reference ranges have not been characterised. This limits genetic studies of disease in African populations as there is a lack of reference data on healthy African individuals. In a sequencing study of ALS in a South African population, none of the genetic candidates investigated were associated with ALS despite them being marked as risk factors in European populations [7]. Taking the repeat range for C9ORF72, the gene most commonly implicated in European ALS populations, as an example, it can be seen that the frequency distribution in healthy populations is not the same in Africans and Europeans (Fig 2).

The same inconsistencies can be seen when looking at other ALS risk genes identified in European populations such as ATXN1 [7]. Evidently, healthy African and European populations have different sized repeat lengths in ALS risk genes. It is thus not surprising that these ALS “risk” genes showed no association with ALS in African ALS populations [7, 8]. This raises the question of whether the associations between STRs and ALS described in European populations are merely spurious associations. Alternatively, they could be population-specific disease modifiers with disease-specific pathways in other populations than requiring identification. This example highlights the problems that arise from attempting to generalise European-specific findings to other populations. Furthermore, the importance of including under-represented populations in research is illustrated.
Why bother and what is to come?
As the research from the field of genetics starts to be applied in health care, there is a risk of under-sequenced populations being left behind. GWAS for disease associations can be used to generate a genetic risk score. While these risk scores are not yet accurate at an individual level, there is hope for their use in the future in informing a precision-based disease approach. Due to differences in LD structure, these risk scores cannot be accurately translated across populations. Failing to generate population-specific risk scores could have adverse health consequences in under-sequenced populations.
An advantage to increasing diversity in genetics research is the uncovering of additional genetic variants. Rare genetic variants are often restricted to a particular population. By expanding sequencing efforts beyond European cohorts, more disease-causing rare variants can be found. These will enhance understanding of disease mechanisms. One more advantage is the creation of a more complete picture of the human genome. A recent sequencing study of 910 individuals of African descent estimated that the pan-African genome contains 10% more DNA than is currently present in the reference human genome [9]. With the origins of humanity lying in Africa [10], African cohorts hold the greatest genetic diversity. Despite the potential this holds for knowledge of the human genome, African populations remain the minority in sequencing studies.
The origins of humanity lie in Africa, so why is this population the minority in sequencing studies?
Fortunately, researchers are fitting more pieces into the human genome puzzle. The Greater Middle East Variome Project is an example of a successful effort to build a genetic database representing Asian populations. Furthermore, in 2012, the Human Hereditary and Health in Africa Consortium (H3Africa) was established to facilitate pan-African genetics research. Numerous success stories have evolved from this initiative. For example, a recent study of Nigerian women with breast cancer identified distinct genetic signatures which could partly explain why in African populations, a more aggressive and fatal form of breast cancer is seen compared to Europeans [11]. The Three Million African Genomes (3MAG) initiative is another promising initiative. This upcoming project headed by the African Society of Human Genetics aims to capture the entire genetic diversity of the African continent [12]. They hope to use this information to build a reference database of African genomes- an encouraging step for important puzzle pieces to fall into place.
The human genome puzzle remains incomplete for now. Many issues need to be addressed before genetics can be said to be representative. Before more attempts are made to uncover increasingly small trait or disease associations in larger and larger European samples, other populations need to be investigated. More initiatives like 3MAG and collaborations to help build infrastructure, gather resources, and increase education are needed. Only when all the puzzle pieces scattered across the globe are collected and assembled, will a coherent genetic picture emerge.
About the author

Kayleigh is a second year Neuroscience master’s student. She has a broad range of interests including neuroscience, genetics and biochemistry. Prior to her studies in Amsterdam she worked with an ALS research group in South Africa. She is especially passionate about psychiatry and how research in this field can be used to improve diagnostic tools and patient outcome. Furthermore, she hopes to contribute to translating science to make it more accessible for the general public.
Further reading
- Lander, S., Linton, L. M., Birren, B., Nusbaum, C., Zody, M. C., Baldwin, J., Devon, K., Dewar, K., Doyle, M., FitzHugh, W., Funke, R., Gage, D., Harris, K., Heaford, A., Howland, J., Kann, L., Lehoczky, J., LeVine, R., McEwan, P., … Yeh, R.-F. (2001). Initial sequencing and analysis of the human genome International Human Genome Sequencing Consortium* The Sanger Centre: Beijing Genomics Institute/Human Genome Center. In Nature, (Vol. 409).
- Uffelmann, E., Huang, Q. Q., Munung, N. S., de Vries, J., Okada, Y., Martin, A. R., Martin, H. C., Lappalainen, T., & Posthuma, D. (2021). Genome-wide association studies. Nature Reviews Methods Primers, 1(1), 59. https://doi.org/10.1038/s43586-021-00056-9
- Rotimi, C. N., & Adeyemo, A. A. (2001). Expanding diversity in genomics. In Nature Biotechnol (Vol. 409, Issue 6).
- Evans, D. M., & Cardon, L. R. (2005). A Comparison of Linkage Disequilibrium Patterns and Estimated Population Recombination Rates across Multiple Populations. In Am. J. Hum. Genet (Vol. 76).
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. (2011). Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron. 72(2):245–56.
- Dashnow H, Lek M, Phipson B, Halman A, Sadedin S, Lonsdale A, et al. (2018). STRetch: Detecting and discovering pathogenic short tandem repeat expansions. Genome Biol. 19(1):1–13.
- Nel, M., Mavundla, T., Gultig, K., Botha, G., Mulder, N., Benatar, M., Wu, J., Cooley, A., Myers, J., Rampersaud, E., Wu, G., & Heckmann, J. M. (2021). Repeats expansions in ATXN2, NOP56, NIPA1 and ATXN1 are not associated with ALS in Africans. IBRO Neuroscience Reports, 10, 130–135. https://doi.org/10.1016/j.ibneur.2021.02.002
- Nel M, Agenbag GM, Henning F, Cross HM, Esterhuizen A, Heckmann JM. (2019). C9orf72 repeat expansions in South Africans with amyotrophic lateral sclerosis. J Neurol Sci 19 [Internet]. 401(March 2019):51–4. Available from: https://doi.org/10.1016/j.jns.2019.04.026
- Sherman, R. M., Forman, J., Antonescu, V., Puiu, D., Daya, M., Rafaels, N., Boorgula, M. P., Chavan, S., Vergara, C., Ortega, V. E., Levin, A. M., Eng, C., Yazdanbakhsh, M., Wilson, J. G., Marrugo, J., Lange, L. A., Williams, L. K., Watson, H., Ware, L. B., … Salzberg, S. L. (2019). Assembly of a pan-genome from deep sequencing of 910 humans of African descent. In Nature Genetics (Vol. 51, Issue 1, pp. 30–35). Nature Publishing Group. https://doi.org/10.1038/s41588-018-0273-y
- J. H. Relethford, “Genetic evidence and the modern human origins debate,” Heredity, vol. 100, no. 6. pp. 555–563, Jun. 23, 2008. doi: 10.1038/hdy.2008.14.
- Ansari-Pour, N., Zheng, Y., Yoshimatsu, T. F., Sanni, A., Ajani, M., Reynier, J. B., Tapinos, A., Pitt, J. J., Dentro, S., Woodard, A., Rajagopal, P. S., Fitzgerald, D., Gruber, A. J., Odetunde, A., Popoola, A., Falusi, A. G., Babalola, C. P., Ogundiran, T., Ibrahim, N., … Olopade, O. I. (2021). Whole-genome analysis of Nigerian patients with breast cancer reveals ethnic-driven somatic evolution and distinct genomic subtypes. Nature Communications, 12(1). https://doi.org/10.1038/s41467-021-27079-w
- Ambroise Wonkam. (2021). Sequence three million genomes across Africa. Nature, (Vol. 590, pp.209-211).
Image credits
Cover photo from https://www.istockphoto.com/nl/foto/world-genetic-dna-strand-gm1356614971-430743095?phrase=genetic%20world%20puzzle; figure 1 adapted by Katelyn Richards from https://www.bcgsc.ca/sites/default/files/Short%20Tandem%20Repeats.jpeg; figure 2 was created in GraphPad Prism by the author as part of her thesis with the Neurology Research group at the University of Cape Town
{kind=link}